Selected works
Survival mediation analysis with the death-truncated mediator: The completeness of the survival mediation parameter
|
Abstract:
In medical research, the development of mediation analysis with a survival outcome has facilitated investigation into causal mechanisms. However, studies have not discussed the death-truncation problem for mediators, the problem being that conventional mediation parameters cannot be well-defined in the presence of a truncated mediator. In the present study, we systematically defined the completeness of causal effects to uncover the gap, in conventional causal definitions, between the survival and nonsurvival settings. We propose a novel approach to redefining natural direct and indirect effects, which are generalized forms of conventional causal effects for survival outcomes. Furthermore, we developed three statistical methods for the binary outcome of survival status and formulated a Cox model for survival time. We performed simulations to demonstrate that the proposed methods are unbiased and robust. We also applied the proposed method to explore the effect of hepatitis C virus infection on mortality, as mediated through hepatitis B viral load. |
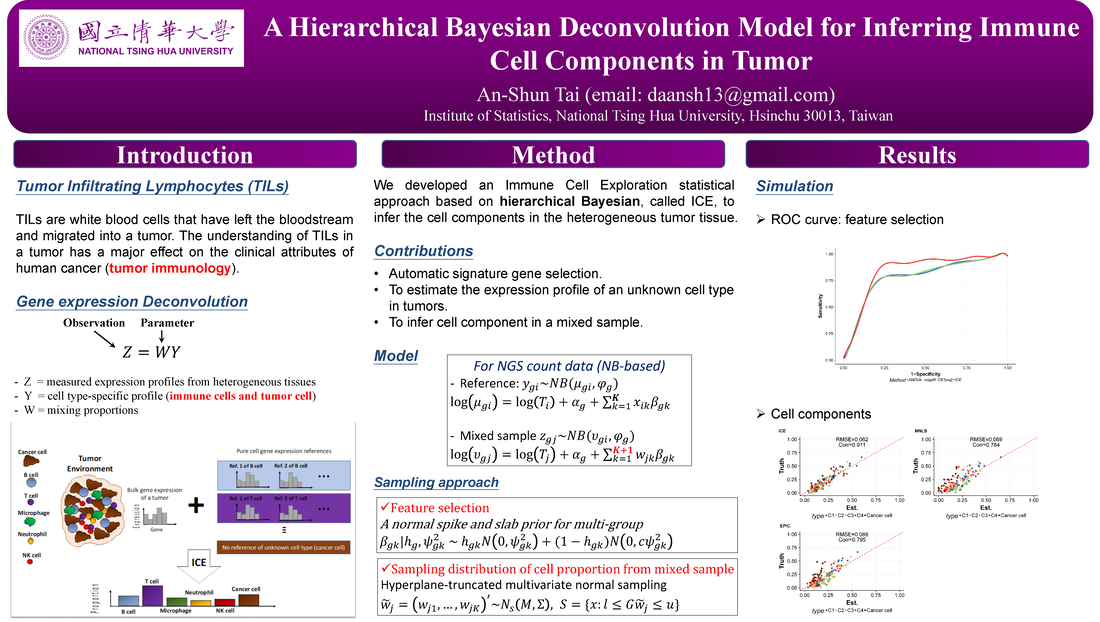
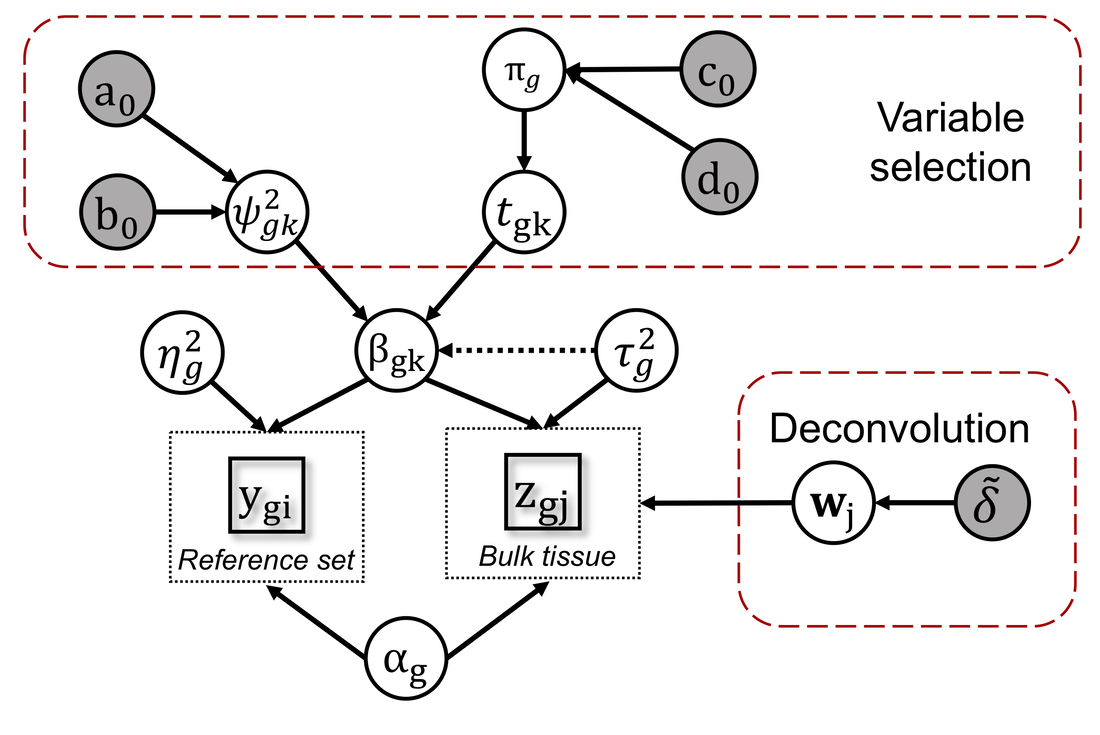
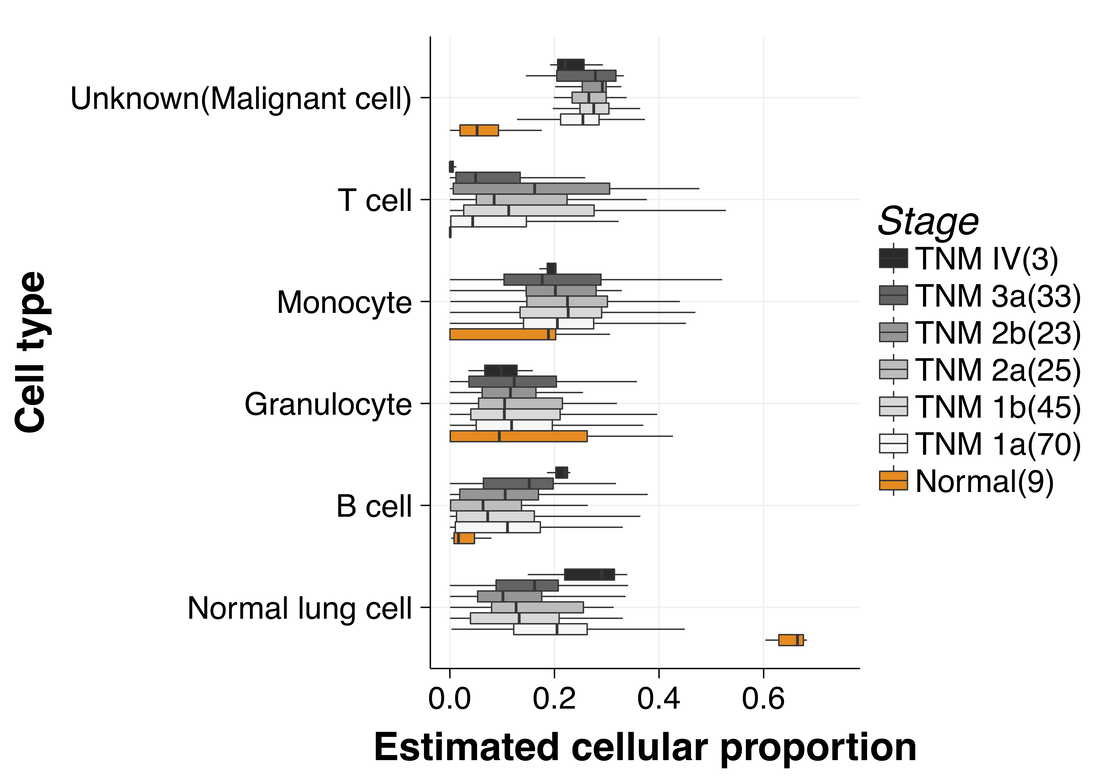
BayICE: A hierarchical Bayesian deconvolution model with stochastic search variable selection
|
Abstract:
Gene expression deconvolution is a powerful tool for exploring the microenvironment of complex tissues comprised of multiple cell groups using transcriptomic data. Characterizing cell activities for a particular condition has been regarded as a primary mission against diseases. For example, cancer immunology aims to clarify the role of the immune system in the progression and development of cancer through analyzing the immune cell components of tumors. To that end, many deconvolution methods have been proposed for inferring cell subpopulations within tissues. Nevertheless, two problems limit the practicality of current approaches. First, all approaches use external purified data to preselect cell type-specific genes that contribute to deconvolution. However, some types of cells cannot be found in purified profiles and the genes specifically over- or under-expressed in them cannot be identified. This is particularly a problem in cancer studies. Hence, a preselection strategy that is independent from deconvolution is inappropriate. The second problem is that existing approaches do not recover the expression profiles of unknown cells present in bulk tissues, which results in biased estimation of unknown cell proportions. Furthermore, it causes the shift-invariant property of deconvolution to fail, which then affects the estimation performance. To address these two problems, we propose a novel deconvolution approach, BayICE, which employs hierarchical Bayesian modeling with stochastic search variable selection. We develop a comprehensive Markov chain Monte Carlo procedure through Gibbs sampling to estimate cell proportions, gene expression profiles, and signature genes. Simulation and validation studies illustrate that BayICE outperforms existing deconvolution approaches in estimating cell proportions. Subsequently, we demonstrate an application of BayICE in the RNA sequencing ofpatients with non-small cell lung cancer. The model is implemented in the R package “BayICE” and the algorithm is available for download. Availability and Implementation: The BayICE R package is available at https://github.com/AshTai/BayICE. |
Decomposing the subclonal structure of tumors with two-way mixture models on copy number aberrations
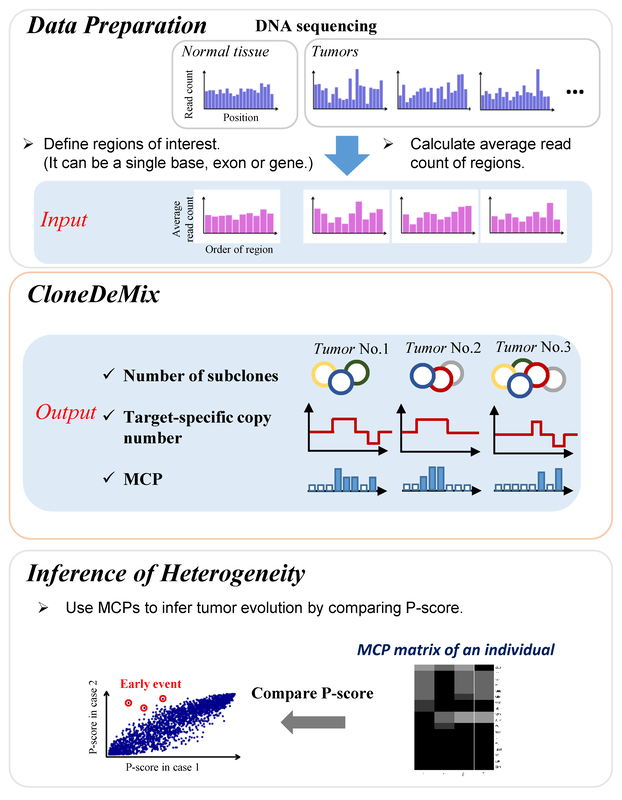
|
Abstract:
Motivation: Multistage tumorigenesis is a dynamic process characterized by the accumulation of mutations. Thus, a tumor mass is composed of genetically divergent cell subclones. With the advancement of next-generation sequencing (NGS), mathematical models have been recently developed to de-mix tumor subclonal architecture among single-nucleotide variants (SNVs) from DNA sequencing data. However, somatic copy number aberrations (CNAs) also play critical roles in carcinogenesis. Therefore, further modeling subclonal CNAs composition would hold the promise to improve the analysis of tumor heterogeneity and cancer evolution. Results: We developed a two-way mixture Poisson model, named CloneDeMix, for the deconvolution of read-depth information and inferred the subclonal copy number of each target region, mutational cellular prevalence (MCP), subclone composition, and the order in which mutations occurred in the evolutionary hierarchy. The performance of CloneDeMix was systematically assessed in simulations. Furthermore, we also demonstrated its applicability using head and neck cancer samples from TCGA. Our results inform about the extent of subclonal CNA diversity, and a group of candidate genes that probably initiate lymph node metastasis during tumor evolution was also discovered. Most importantly, these driver genes located at 11q13.3 which is highly susceptible to copy number change in head and neck cancer genomes. Briefly, this framework has implications for improved modeling of tumor evolution and the importance of inclusion of subclonal CNAs. Availability and Implementation: The CloneDeMix R package is available at https://github.com/AshTai/CloneDeMix. |